

“Aging can be defined as the time-related deterioration of the physiological functions necessary for survival and fertility.”
From Developmental Biology, 6th edition (Gilbert, 2000)
“At the biological level, aging results from the impact of the accumulation of a wide variety of molecular and cellular damage over time. This leads to a gradual decrease in physical and mental capacity, a growing risk of disease and ultimately death.”
From WHO (https://www.who.int/news-room/fact-sheets/detail/ageing-and-health)
“Aging is the process of becoming older. In humans, aging represents the accumulation of changes in a human being over time and can encompass physical, psychological, and social changes. Aging increases the risk of human diseases such as cancer, Alzheimer’s disease, diabetes, cardiovascular disease, stroke and many more.”
From Wikipedia (https://en.wikipedia.org/wiki/Ageing):
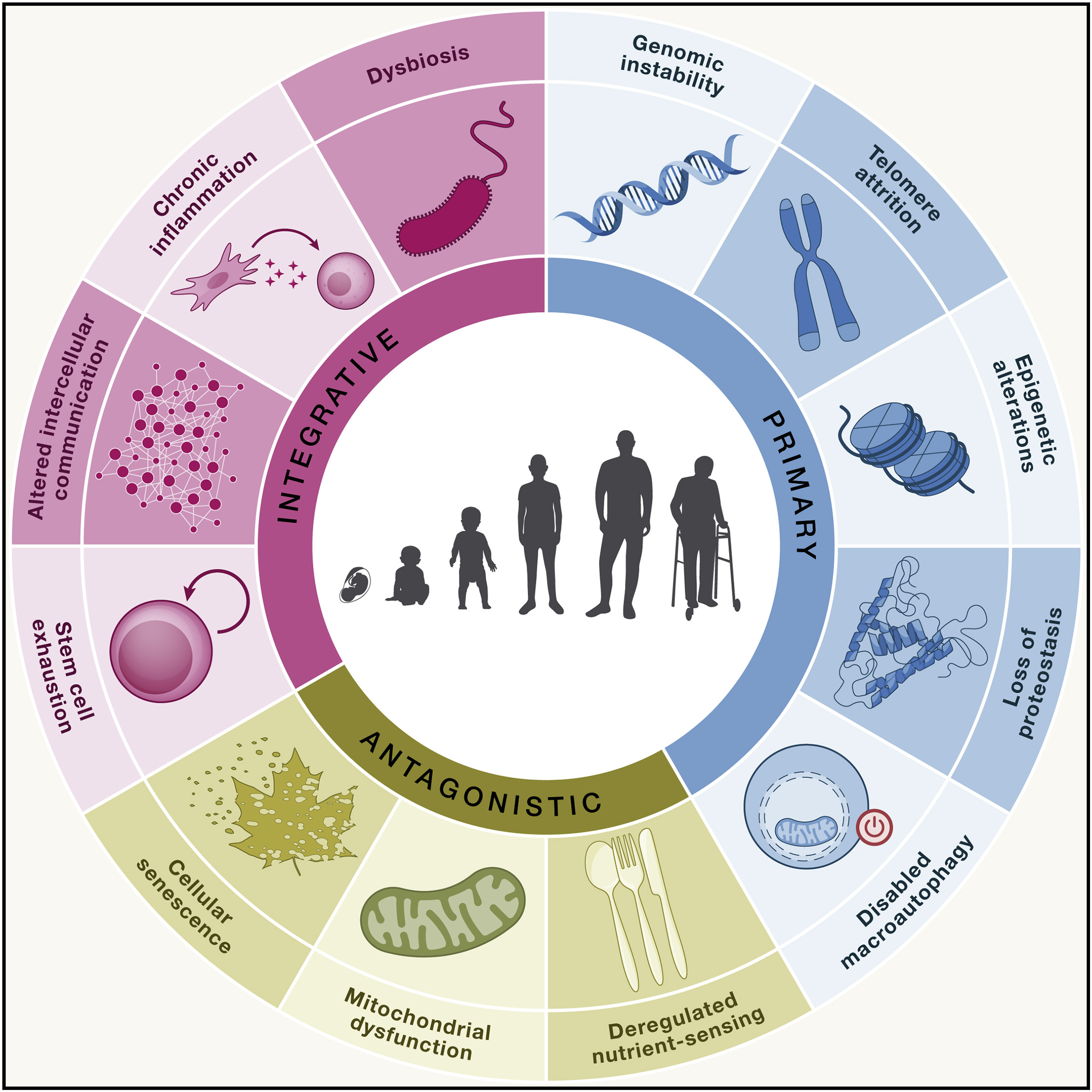
The distinction among ‘‘hallmarks’’ is intrinsically diffuse, since they interact and are not independent of each other. Therefore, their classification is inevitably arbitrary. It was proposed that the following three criteria must apply for each hallmark of aging (López-Otín et al., 2023):
The following are the latest list of 12 aging hallmarks (López-Otín et al., 2023):
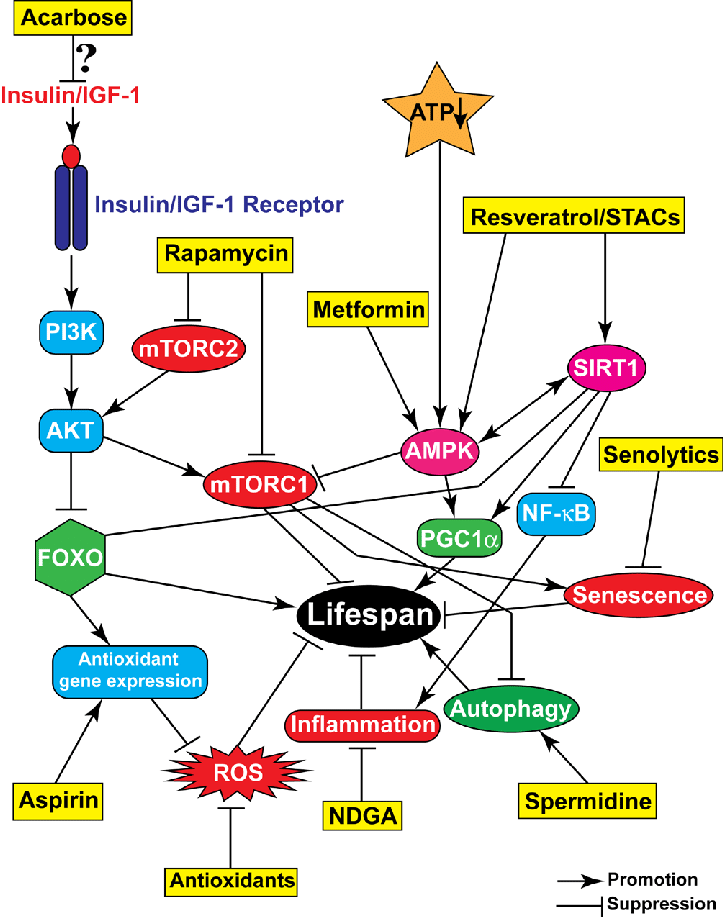
The twelve hallmarks of aging collectively encompass the major molecular, cellular, and systemic processes that drive organismal aging. Over the past several decades, extensive efforts worldwide have sought to develop interventions targeting each of these hallmarks with the goal of delaying aging, improving healthspan, or the prevention/treatment of age-related diseases. Below are summarized representative examples of experimental and preclinical interventions corresponding to each hallmark.

Despite decades of intensive research, aging remains largely refractory to effective intervention. Numerous hallmarks of aging and corresponding therapeutic strategies have been proposed and implemented. While such interventions can delay or modulate specific aspects of aging, none have been shown to halt the aging process in complex organisms.
A central limitation of prevailing approaches is that they conceptualize aging as a collection of independent molecular damage across distinct pathways, rather than as the failure of a higher-order, coordinated biological program. In doing so, they overlook a fundamental biological reality: robust and reproducible rejuvenation already exists in nature
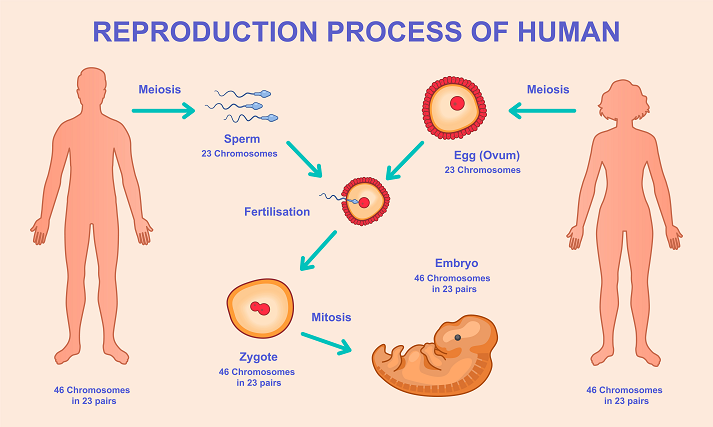
In all sexually reproducing organisms, a profound rejuvenation event occurs during reproduction. The fusion of sperm and oocyte gives rise to a zygote that is biologically young, despite originating from aged parental cells. During early embryogenesis, accumulated age-associated epigenetic marks are erased, transcriptional noise is reduced, and cellular identity undergoes global reprogramming. Recent studies have demonstrated that epigenetic aging clocks reset to near zero during this developmental window, effectively restoring a youthful cellular state (Yang et al., 2023). Consequently, offspring inherit their parents’ genetic information while remaining largely free of the cellular aging burden accumulated across the parental lifespan, thereby ensuring species continuity across generations.
This observation raises a fundamental question: if a highly coordinated and robust rejuvenation program exists and is repeatedly executed with remarkable fidelity during reproduction, why is it not activated in somatic tissues as organisms age?
One plausible explanation lies in evolutionary optimization. Natural selection acts primarily to maximize reproductive success and species persistence, rather than individual longevity. Under conditions of finite resources and persistent environmental risk, deploying powerful rejuvenation mechanisms broadly in somatic tissues may confer limited evolutionary advantage while increasing the risk of adverse outcomes such as uncontrolled proliferation or tumorigenesis. As a result, rejuvenation programs appear to be developmentally and epigenetically gated, restricted to the germline-to-zygote transition where they most effectively enhance fitness at the species level.
From this perspective, aging is not solely the consequence of cumulative molecular damage, but also the result of an evolutionarily enforced suppression of a robust rejuvenation program in somatic cells. This framework suggests that meaningful intervention in aging may require more than repairing individual forms of damage; it may necessitate the controlled and context-dependent reactivation of key elements of the embryonic rejuvenation program.
Accordingly, elucidating how nature achieves rejuvenation during reproduction, how this rejuvenation program is subsequently silenced, and how it might be safely and precisely reactivated in somatic tissues represents one of the most promising avenues toward transformative interventions in aging.
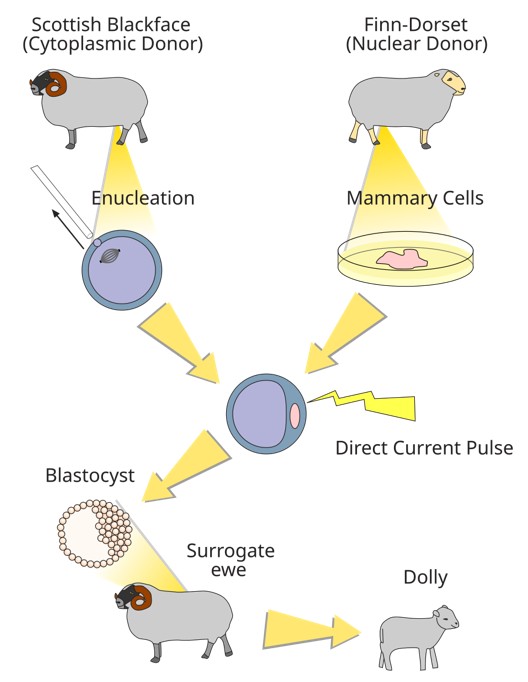
The journey toward rejuvenation began in the early 1960s when John Gurdon and his collaborators discovered animal cloning in frogs (Gurdon, 1962). Three decades later, in 1996, mammalian cloning was achieved with the birth of Dolly the sheep (Campbell et al., 1996), followed by the cloning of other mammalian species. These breakthroughs demonstrated that the cytoplasm of a mature oocyte contains molecules capable of reprogramming a somatic nucleus into an embryonic state, enabling the development of a new organism. Scientists hypothesized that the oocyte’s cytoplasm must contain a complex constellation of reprogramming factors essential for resetting cellular age. But what are those factors in the oocyte’s cytoplasm that caused the reprogramming?
Humans have approximately 20,000 protein-coding genes, but the total number of genes, including non-coding RNA genes, is much higher, with some estimates exceeding 46,000 (Amaral et al., 2023). Even when restricted to protein-coding genes, the number of possible combinations involving two to six genes out of approximately 20,000 amounts to 8.88 × 10²². Such an astronomical number far exceeds the capacity of current experimental or computational approaches to be tested exhaustively. What makes this task more complicated is that some genes in a combination need to be over expressed, and some need to be inhibited, and sometime need to be spatially and timely regulated.
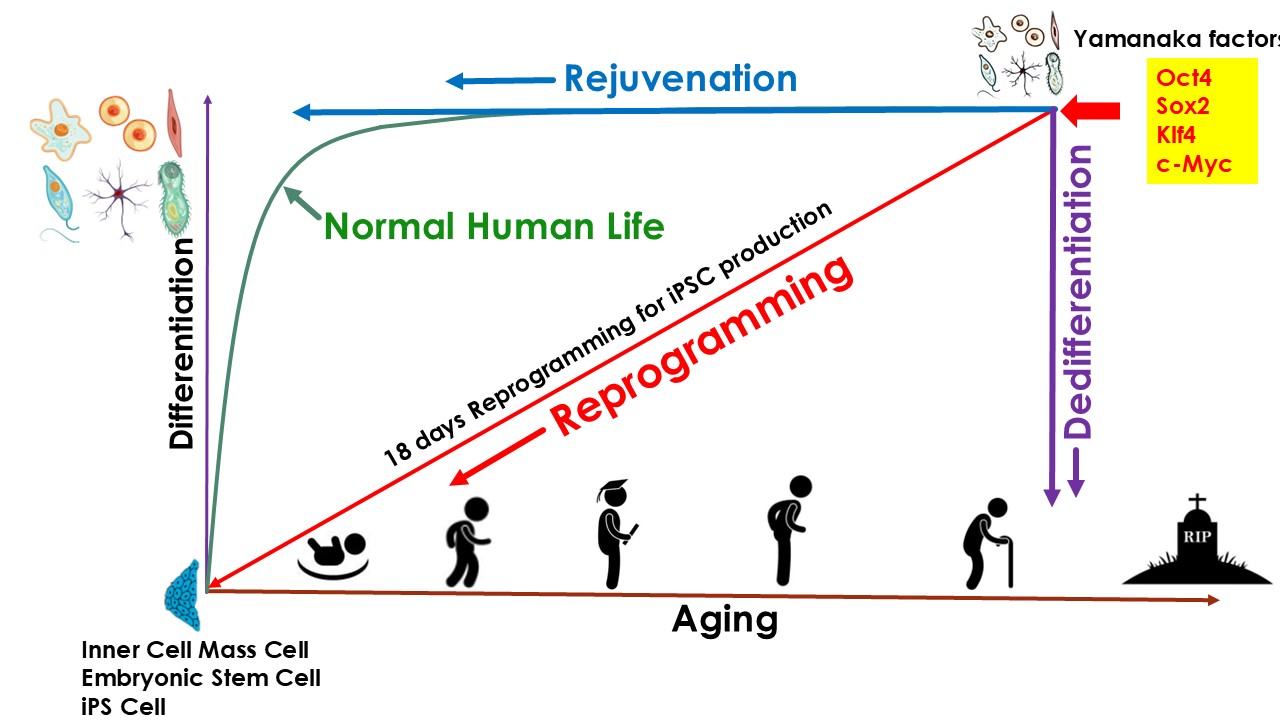
Thanks to the discovery of induced pluripotent stem cell (iPSC) technologies in 2006 by Yamanaka (Takahashi & Yamanaka, 2006), who, because of it, got Nobel Prize in 2012, scientists know that only 4 genes (Oct4, Klf4, Sox, and Myc) (OKSM), so called Yamanaka factors, can reprogram somatic cells into iPSC cells. iPSC reprogramming represents the most robust experimentally validated example of cellular rejuvenation by using defined factors. Multiple studies have shown that reprogramming aged or senescent somatic cells to pluripotency effectively resets epigenetic age, restores youthful transcriptional programs, and reverses several hallmarks of cellular aging. Though iPSC reprogramming successfully rejuvenated cells, it also caused dedifferentiation of the cells, thus the cells lost their original phenotype. In other words, the rejuvenation and differentiation are coupled during the iPSC reprogramming process. As a result, iPSC technologies cannot be used directly in the body for rejuvenation purposes, because it could cause teratoma or other type of tumors in the body (Liu, 2008). However, there are still a lot of achievements on rejuvenation applications based on iPSC technology. One of the most important achievements is called Partial Reprogramming:
This is to initiate reprogramming with OSKM, interrupt the process at an intermediate state, and allow cells to return to their original identity. This transient cellular perturbation, known as “partial” reprogramming, is able to rejuvenate cellular markers of aging such as the DNA methylation clock, DNA damage, epigenetic patterns, and aging-associated changes in the transcriptome, both in vitro and in vivo. Upon interruption of partial reprogramming, cells re-stablish their original epigenetic and transcriptional status in a process of re-differentiation that, interestingly, does not re-stablish the erased aging-associated changes and therefore resets the epigenome and transcriptome to a younger state. (Browder et al., 2022; Ocampo et al., 2016; Olova et al., 2019). Various in vivo, in vitro studies have explored partial reprogramming strategies, including approaches based on transient mRNA delivery or tightly controlled expression of canonical reprogramming factors. These studies demonstrate that partial reprogramming can induce certain rejuvenative effects while avoiding full pluripotency.
However, in most existing approaches, the extent of reprogramming is regulated primarily through temporal control—limiting the duration of factor exposure to achieve rejuvenation while minimizing dedifferentiation. As such, these strategies do not explicitly aim to mechanistically decouple rejuvenation from dedifferentiation during the reprogramming process. Instead, they rely on incomplete progression along a coupled reprogramming trajectory, leaving the fundamental linkage between rejuvenation and loss of cell identity largely unaddressed.
As mentioned above, the rejuvenation during iPSc reprogramming is linked to loss of somatic cell identity. Consequently, canonical iPSC-based approaches cannot be directly applied in vivo for therapeutic rejuvenation.
Despite this limitation, iPSC technology provides a powerful and informative starting point. Rather than viewing dedifferentiation as an unavoidable consequence of rejuvenation, we believe they can be separated or decoupled.
A key biological observation supports the conceptual separability of these processes. In humans, a fertilized zygote develops into a fully differentiated neonate within approximately 40 weeks, generating nearly all somatic cell types of the adult organism. During this developmental interval, extensive cellular differentiation occurs, yet biological aging has scarcely begun. This temporal dissociation suggests that cellular differentiation and biological aging are governed by largely independent biological programs. If differentiation can proceed without aging during development, then rejuvenation need not be intrinsically linked to dedifferentiation.
This raises a fundamental question: why are rejuvenation and dedifferentiation tightly coupled during iPSC reprogramming, despite being separable during natural development?
We hypothesize that the coupling between rejuvenation and dedifferentiation observed during induced pluripotent stem cell (iPSC) reprogramming is not a biological necessity, but rather a consequence of the pleiotropic and upstream positioning of canonical reprogramming factors within the gene regulatory network. The Yamanaka factors (OCT4, SOX2, KLF4, and c-MYC), together with additional pluripotency-associated regulators(Yu et al., 2007), function as high-level transcriptional and epigenetic remodelers. They bind to thousands of genomic loci, recruit chromatin-modifying complexes, alter histone modifications (e.g., H3K27ac, H3K4me3), promote widespread chromatin accessibility changes, and reorganize three-dimensional genome architecture. These activities collectively reset transcriptional programs governing cell identity.
Importantly, many age-associated molecular features—such as epigenetic drift, altered DNA methylation patterns, transcriptional noise, heterochromatin loss, impaired DNA damage response, and metabolic rewiring—are embedded within the same chromatin and transcriptional landscape that defines somatic cell identity. Thus, global chromatin remodeling during reprogramming simultaneously erases age-associated epigenetic marks and restores youthful gene expression patterns. In this context, rejuvenation arises as a systems-level consequence of large-scale regulatory resetting, rather than as a process intrinsically dependent on dedifferentiation.
We therefore propose that the reprogramming gene regulatory network contains at least two partially overlapping but mechanistically distinguishable modules. The first module governs dedifferentiation and pluripotency acquisition, including suppression of lineage-specific transcription factors, activation of pluripotency circuits, and reconfiguration of enhancer–promoter interactions characteristic of embryonic stem cells. The second module governs rejuvenation, encompassing restoration of youthful DNA methylation states, re-establishment of heterochromatin structure, reduction of transcriptional entropy, enhancement of DNA repair capacity, normalization of mitochondrial and metabolic programs, and attenuation of senescence-associated signaling pathways.
Canonical reprogramming factors likely reside at the apex of this hierarchical architecture, simultaneously activating both modules due to their broad chromatin-opening and pioneer factor activities. We posit that these modules can be disentangled at the level of downstream effectors. By systematically mapping transcription factor binding hierarchies, epigenetic remodeling trajectories, and temporal gene expression dynamics—using single-cell multi-omics and network inference approaches—it should be possible to identify and selectively activate regulatory nodes that drive rejuvenation-specific processes while avoiding activation of core pluripotency circuitry.
Such selective modulation would enable restoration of youthful molecular and functional states without erasure of somatic identity. This modular and mechanistically grounded framework constitutes the conceptual foundation of our strategy to decouple rejuvenation from dedifferentiation.
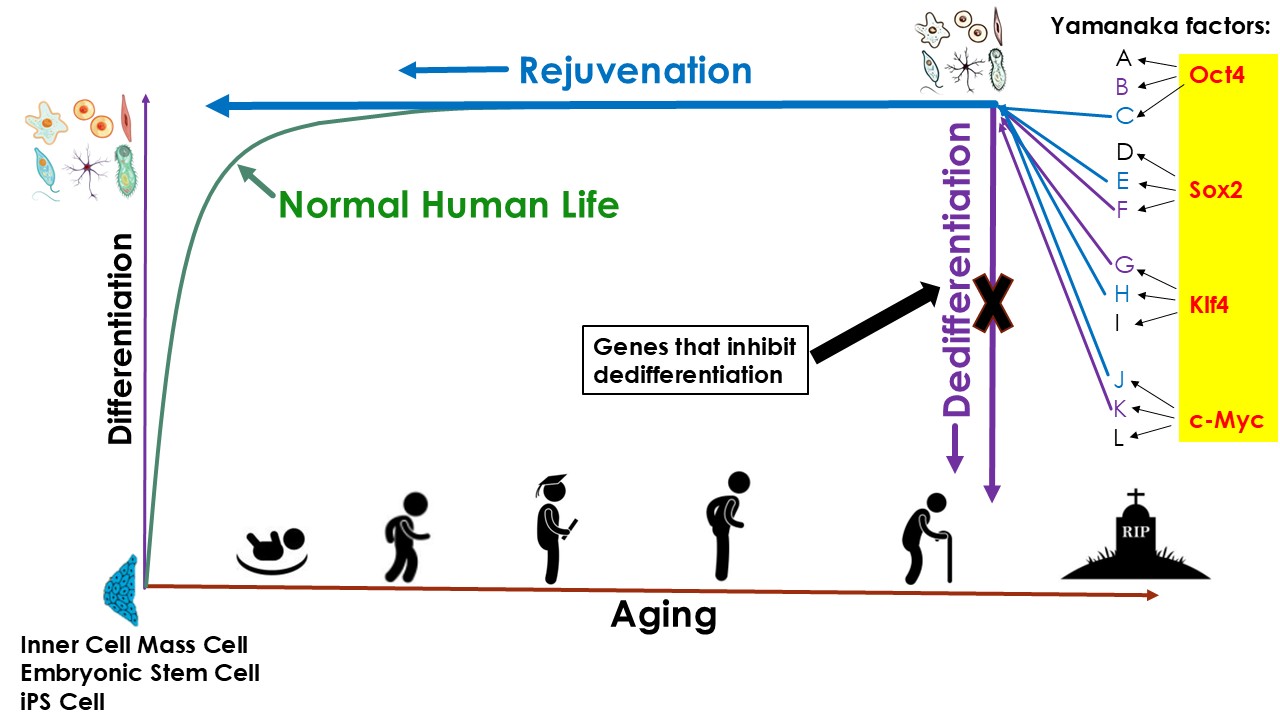
Given that iPSC reprogramming can robustly rejuvenate cells, we adopted a rational, mechanism-driven strategy: rather than attempting to identify rejuvenation-inducing gene combinations de novo, we sought to deconstruct the reprogramming process itself. Specifically, we aimed to identify genetic perturbations that suppress dedifferentiation while allowing rejuvenation to proceed during reprogramming.
We first sought to identify genes associated primarily with cellular differentiation status. To this end, we analyzed publicly available transcriptomic datasets. Using advanced bioinformatics and data science methodologies, we identified genes exhibiting strong differential expression between young somatic cells and early embryonic or pluripotent cells. These genes fell into two principal categories:
Through this analysis, we identified 527 differentiation-related genes with strong expression bias toward either somatic or pluripotent states.
In parallel, we applied the same analytical framework to gene expression data obtained during iPSC reprogramming, enabling the identification of genes differentially regulated between somatic cells and iPSCs. Because reprogramming intrinsically encompasses both dedifferentiation and rejuvenation, genes altered during this process are expected to participate in differentiation-related pathways, aging-associated processes, or both. This analysis yielded 982 reprogramming-associated genes.
To further isolate aging-specific components, we independently analyzed publicly available aging-related transcriptomic datasets, identifying 459 aging-associated genes.
As expected, substantial overlap was observed among genes associated with differentiation, reprogramming, and aging. This overlap is biologically plausible, as senescent cells are typically terminally differentiated. Following integrative analysis and filtering, we selected approximately 200 candidate genes that were strongly associated with differentiation while showing minimal association with aging-related expression changes. These genes were prioritized for downstream functional screening.
To effectively identify novel rejuvenation methods, it is essential to first establish a reliable and high-throughput screening platform dedicated to evaluating cellular rejuvenation.
Aging is believed to commence as early as embryonic development (Gallardo et al., 2025), reflecting intrinsic biological programs that accompany cellular differentiation and tissue formation. Nevertheless, the pathophysiological manifestations of aging—such as functional decline and age-associated diseases—predominantly arise during adulthood. Accordingly, our rejuvenation research centers on adult cellular aging (cellular senescence).
Among the various methods for inducing cellular senescence, replicative senescence is the closest to natural cellular aging, as it occurs naturally in cells that divide over time, particularly in actively proliferating cells. This process closely mimics the gradual decline in cellular function observed during organismal aging, making it a suitable model for studying cellular aging (Ahn et al., 2025). Here we employ adult human arterial endothelial cells (HAECs) as an experimental model. There are two major reasons that we chose HAEC. One is HAECs from the same location in an artery are relatively homogeneous compared to other types of human primary cells. They have unique shapes in cell culture as well as unique surface markers, making it easier to identify and quantify. In addition, because of the relative homogeneousness, it would be easier to identify the minor shift on phenotype changes as well as genes expression profile changes. Another reason we chose HAECs is that endothelial cells are known to be one of the first cell types to become senescent with advancing age (Bloom et al., 2023; Grosse et al., 2020). In vivo, endothelial senescence occurs in multiple vascular beds including kidney (Cohen et al., 2021), retina (Shosha et al., 2018), liver (Grosse et al., 2020), brain (Kiss et al., 2020) and aorta (Yokoi et al., 2006), suggesting that endothelial senescence contributes to a variety of pathological processes associated with vascular dysfunction. Therefore, rejuvenating endothelial cells holds significant therapeutic potential for preventing and treating many major diseases associated with vascular dysfunction.
We have built a propriety rejuvenation screening platform using HAECs (US patent pending, Application# 63/960,217) for the high throughput screening of rejuvenation gene cocktails. In this screening model, we use replicative aging human artery endothelial cells (HAEC) (non-growing) as starting cells and use their growth capability as the first parameter of rejuvenation, followed by identity verification and other parameters, as well as RNA sequencing analysis.
To functionally interrogate the role of our differentiation-related candidate genes, we incorporated them into the above-mentioned rejuvenation screening platform undergoing reprogramming. Specifically, during reprogramming treatment, we applied gene perturbations in two complementary modes:
By systematically applying these genes during reprogramming, we screened cells exhibited molecular and functional markers of rejuvenation while retaining endothelial cell identity.
Through the above approaches, we identified a set of genes whose perturbation selectively suppresses dedifferentiation without impairing rejuvenation during the reprogramming of senescent HAECs. Under these conditions, cells displayed rejuvenated molecular signatures while maintaining their native endothelial phenotype (Figure 1 – Figure 5).
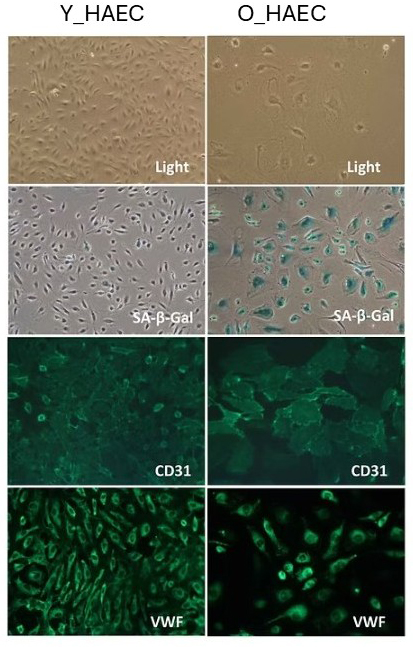
Figure 1: Characterization of young (Y_HAEC) and senescent (O_HAEC) HAECs.
As expected, Y_HAECs are small and exhibit typical endothelial morphology under a light microscope. They are β-Gal negative, indicating that they are not senescent, as confirmed by their continuous growth capability. Additionally, they are CD31 and VWF positive, confirming their endothelial identity.
In contrast, O_HAECs appear larger while retaining endothelial morphology under a light microscope. However, they are β-Gal positive, signifying cellular senescence, as further evidenced by their lack of proliferative capacity. Despite senescence, these cells remain CD31 and VWF positive, confirming that they still maintain their endothelial identity.
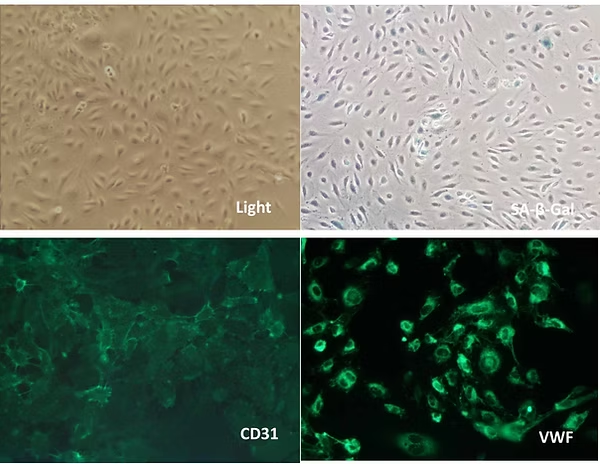
Figure 2. Rejuvenation of Senescent HAECs after treatment.
Following treatment, the treated senescent HAECs (T2_HAEC) exhibit morphology similar to young endothelial cells under a light microscope. They are β-Gal negative, indicating the loss of senescence, and their restored growth capability confirms their rejuvenated state.
Additionally, the T2_HAEC remain CD31 and VWF positive, demonstrating that they have retained their endothelial identity. These findings confirm that our gene combination successfully rejuvenates senescent HAECs while preserving their original cell phenotype.
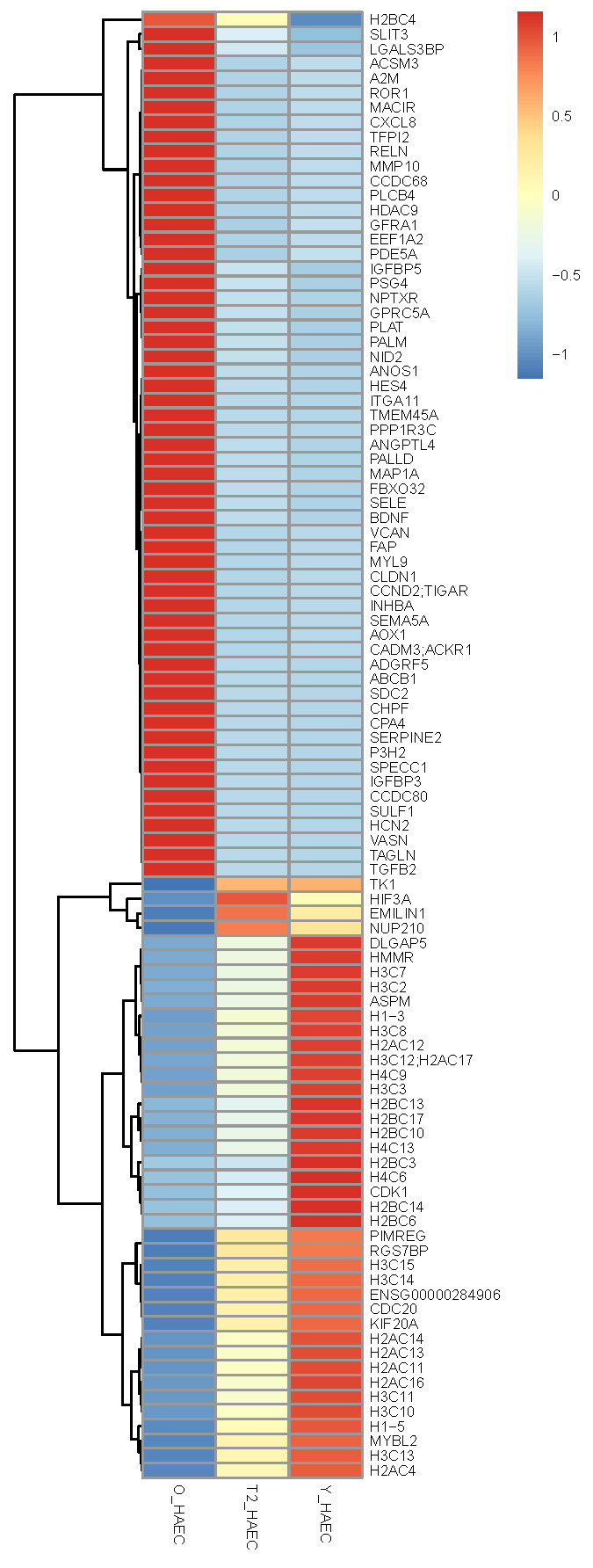
Figure 3. RNA sequencing (RNA-Seq) heatmap of the Old (O_HAEC), Treated (T2_HAEC), and Young (Y_HAEC). The heatmap displays gene expression (z-score normalized) from blue (low) to red (high) across ~110 strongly regulated genes.
Old cells (O_HAEC) show high expression of: Fibrosis / ECM remodeling genes such as VCAN, FAP, COL, ECM-related genes (SULF1, CHPF), TAGLN, MYL9 (smooth muscle / contractile markers), SERPINE2, ANGPTL4, PALM, IGFBP3, NID2, ITGA11; SASP / Inflammatory genes, such as CXCL8, RELN, MMP10, SELE; Pro-aging regulators such as HDAC9 (known aging marker) and PPP1R3C and PALLD
Young HAECs (Y_HAEC) show high expression of: Cell Cycle / Proliferation / Mitosis genes, such as CDK1, CDC20, KIF20A, DLGAP5, HMMR, ASPM, MYBL2, TK1, HIF3A, NUP210, PIMREG, RGS7BP; Histone and chromatin genes (replicative youth signature) such as H2BC, H3C, H4C, H2AC, multiple variants, CENP-A related histones.
Treatment Signature (T2_HAEC) shows that treatment reduces Old-like genes: treatment cells show blue (downregulation) for many Old-high genes: SASP genes drop (CXCL8, MMP10, SELE); ECM/fibrosis markers drop (VCAN, FAP, TAGLN); Aging regulators drop (HDAC9, IGFBP3, SERPINE2). This is direct partial reversal of aging.
Treatment partially restores Young-like genes: in the lower half of the heatmap, many Young-high cell-cycle and chromatin genes that are suppressed in Old become yellow-red (re-activated) in Treatment: CDK1, KIF20A, DLGAP5, CDC20, Histones (H2BC, H3C, H4C), ASPM, MYBL2, H3C10, H1-5, etc.
The heatmap analysis reveals a clear transcriptional trajectory from Young (Y_HAEC) to Old (O_HAEC) HAECs, with the rejuvenation treatment shifting the aged cells back toward a youthful state. Old cells exhibit high expression of pro-inflammatory, ECM-remodeling, and senescence-associated genes (e.g., CXCL8, SERPINE2, VCAN, TAGLN), coupled with broad suppression of cell cycle regulators and chromatin/histone genes. In contrast, young cells show robust activity of DNA replication, mitotic, and chromatin-remodeling programs.
Treated cells (T2_HAEC) demonstrate marked downregulation of aging-associated genes and restoration of youthful gene programs, particularly across cell cycle, chromatin, histone clusters, and mitotic regulators (CDK1, KIF20A, ASPM, HMMR, histone families). Hierarchical clustering positions T2_HAEC cells intermediate between Old and Young, but are biologically closer to Young than Old, indicating substantial yet incomplete rejuvenation—consistent with a controlled, non-reprogramming reversal of endothelial aging.
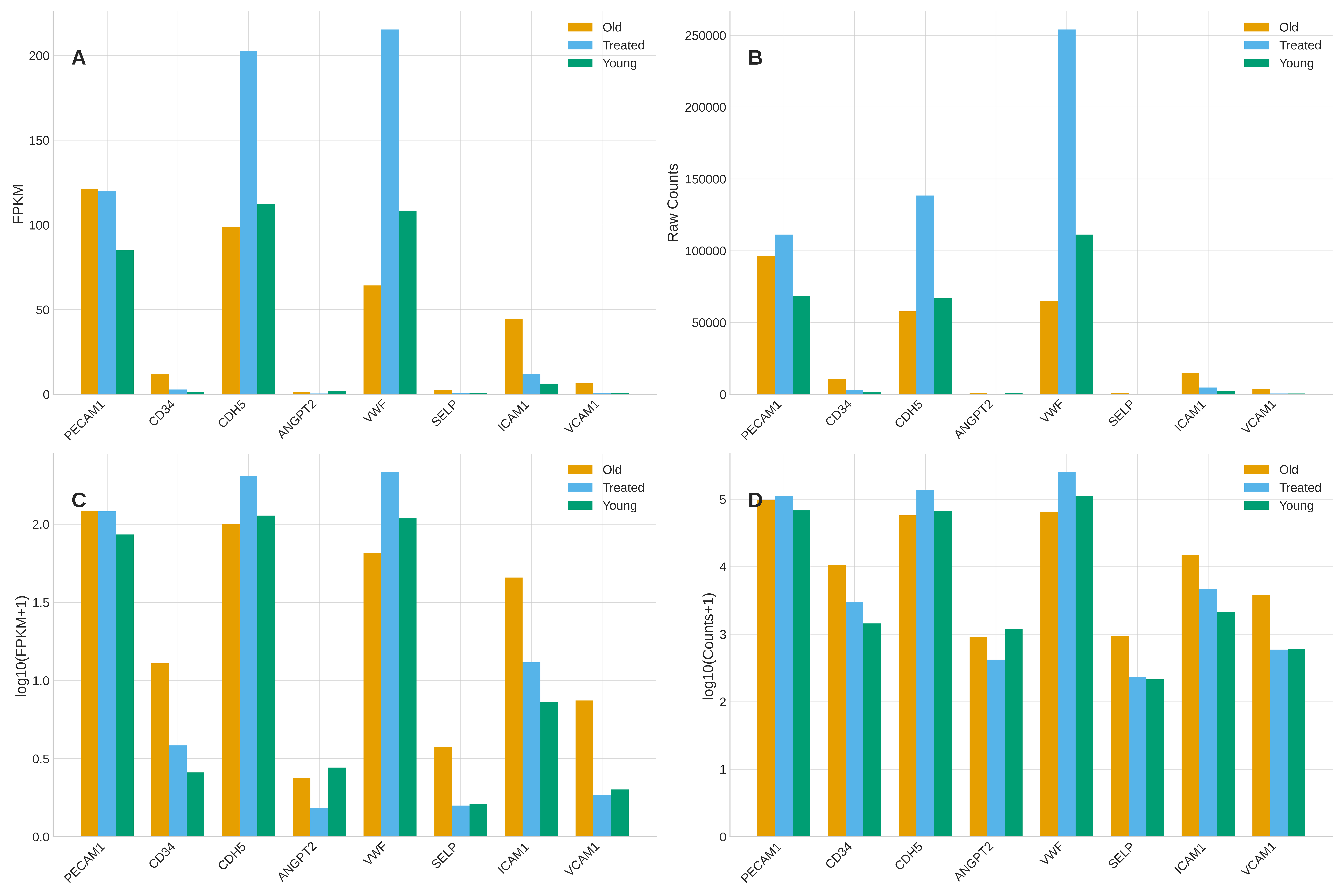
Figure 4. The expression of endothelia markers. Young (Y_HAEC), Old (O_HAEC), and Rejuvenated (T_HAEC). Expression levels of eight endothelial marker and activation-related genes are shown across Young (Y_HAEC), Old (O_HAEC), and Rejuvenated (T_HAEC) human aortic endothelial cells. Panel A displays FPKM expression values; Panel B shows raw RNA-seq counts; Panels C and D present the same data on a log10 scale for clearer visualization across expression ranges. Across all metrics, canonical endothelial identity genes — PECAM1, CD34, CDH5, ANGPT2, VWF, SELP, ICAM1, and VCAM1 — remain similar robust expression among all three groups, including rejuvenated cells, indicating preservation of endothelial lineage.
The endothelial marker gene expression analysis demonstrates that young, senescent, and rejuvenated HAECs all maintain the core endothelial gene expression program, confirming that the rejuvenation intervention does not cause dedifferentiation or loss of vascular identity.
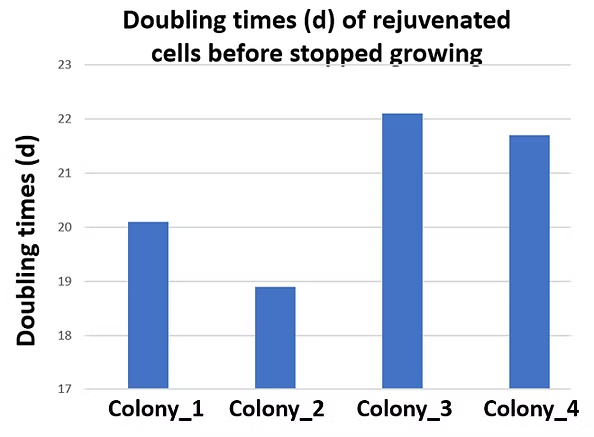
Figure 5. Doubling Times (n) of Rejuvenated HAECs Before Growth Cessation
The rejuvenated HAECs treated with our gene combination were continuously cultured, and their doubling times (d) were calculated. The rejuvenated endothelial cells exhibited doubling times of approximately 22–24 before reaching senescence again.
The initial rejuvenation screening was conducted on senescent HAECs (O_HAEC, also pX+d12.97), where the senescent cells had an unknown passage number (pX) with an additional 12.97 doublings (d12.97). After rejuvenation, the cells regained proliferative capability, completing an additional 22–24 doublings before ceasing growth once more.
These results demonstrate that rejuvenated cells retain a limited proliferative capacity, confirming that they are non-tumorigenic and do not exhibit uncontrolled growth.
Previous studies have suggested the possibility of rejuvenating cells without inducing full dedifferentiation (de Lima Camillo et al., 2025). However, the specific genes or gene combinations responsible for this effect were not disclosed, limiting reproducibility and mechanistic interpretation. By contrast, our work provides, to our knowledge, the first experimentally validated and molecularly defined demonstration of gene-mediated rejuvenation that preserves native somatic cell identity (U.S. Patent Application No. 63/960,217).
Collectively, these findings support the central hypothesis that rejuvenation and dedifferentiation are separable biological processes governed by partially independent regulatory networks. More broadly, they suggest that effective rejuvenation strategies may be achieved not by repairing individual aging hallmarks in isolation, but by selectively reactivating components of an evolutionarily conserved rejuvenation program while actively constraining cell identity loss.
Milestone 1 demonstrated that we can rejuvenate senescent human endothelial cells while preserving their cellular identity by strategically inhibiting dedifferentiation during reprogramming. However, the gene cocktails used at this stage were intentionally broad (including all the Yamanaka Factors) and relied on upstream regulators with wide-ranging chromatin and transcriptional effects. While effective as proof-of-concept, these formulations are not optimized for clinical translation, as they activate multiple regulatory pathways beyond those strictly required for rejuvenation.
Milestone 2 marks a strategic shift from empirical factor combinations to precision regulatory engineering. Our objective is to identify the minimal set of downstream regulatory nodes that are necessary and sufficient to restore youthful cellular function—without activating pluripotency programs. To achieve this, we are combining two core assets:
This integrated platform enables us to map the regulatory architecture underlying rejuvenation at systems scale, identify master control genes, and computationally screen simplified gene combinations before experimental validation. In doing so, Milestone 2 transforms our approach from broad reprogramming-based intervention to rational, mechanism-guided design of optimized rejuvenation gene cocktails with improved safety, specificity, and translational potential.
Although numerous public gene expression datasets on aging, differentiation, or reprogramming are available, their inherent variability—arising from differences in experimental design, biological sources, control selection, and data quality—has led to inconsistent and sometimes contradictory conclusions. Most importantly, until recently, there have been no rejuvenation gene expression datasets available. This is primarily due to the absence of reliable experimental models capable of rejuvenating senescent human cells while preserving their original cellular identity. Consequently, identifying the key regulatory genes that drive human aging and rejuvenation has remained a major challenge.
To address these limitations, there is an urgent need for a comprehensive, time-resolved map of gene expression dynamics encompassing the full course of cellular aging and rejuvenation within the same experimental system.
Building upon the achievements of Milestone 1, we are now positioned to establish the Longitudinal Human Aging and Rejuvenation Gene Expression Atlas, a systematic and high-resolution experimental framework to capture the time-resolved gene expression dynamics across cellular aging, and, critically, rejuvenation under different gene perturbation conditions in senescent HAECs, as well as related differentiation and reprogramming trajectories.
The Longitudinal Human Aging and Rejuvenation Gene Expression Atlas contain the following four core components
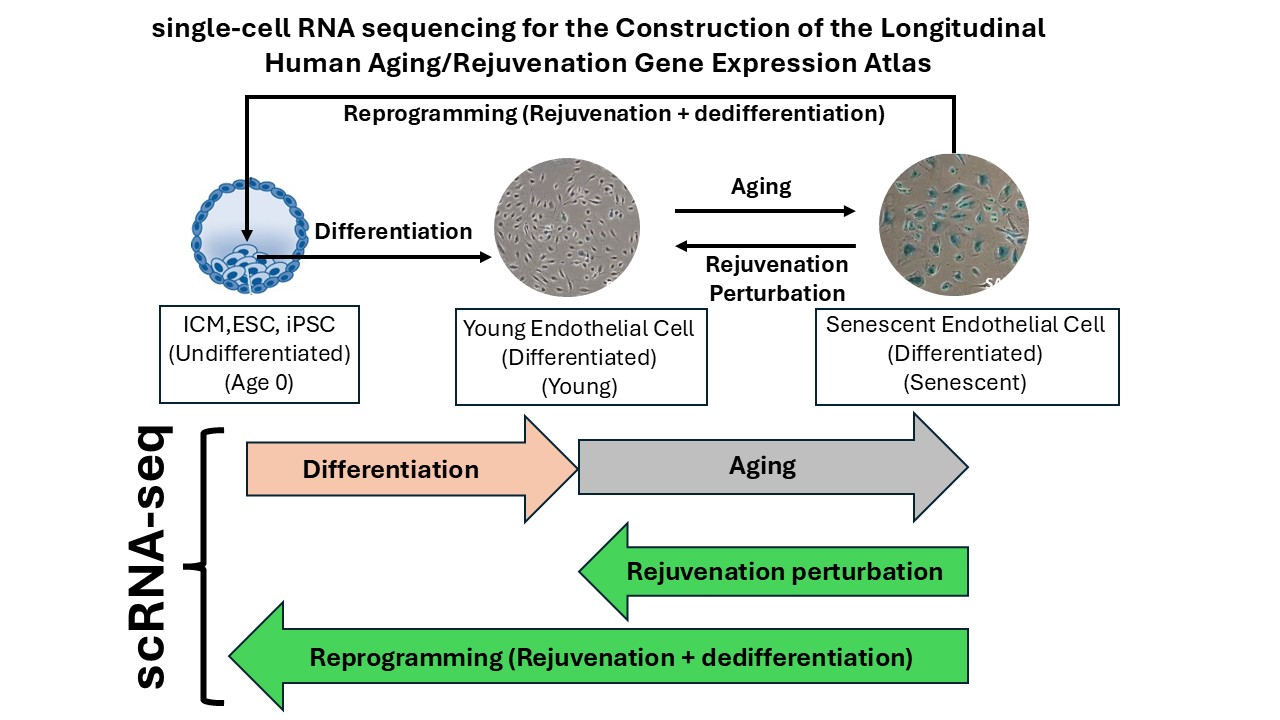
Critically, all datasets are derived from the same primary human cell type, adult HAECs. This single-cell-type design eliminates cross-lineage variability and maximizes our ability to detect subtle, temporally resolved regulatory changes that would be obscured in heterogeneous datasets.
Moreover, all datasets include high-quality supervised labels, such as treatment conditions, cell-cycle and doubling-time metadata, and defined cellular states. These rich annotations substantially increase the value of the atlas for downstream machine-learning applications, particularly for supervised and semi-supervised model fine-tuning.
This integrated atlas will represent the first complete, time-resolved Longitudinal Human Aging and Rejuvenation Gene Expression Atlas. It will serve as a unique, comprehensive resource that captures the regulatory dynamics underlying human cellular aging and rejuvenation. The atlas will also form the foundation for later phases of our AI-driven approaches.
By analyzing the Longitudinal Human Aging/Rejuvenation Gene Expression Atlas, using AI technology, we will identify gene expression signatures corresponding to different days of continuous HAEC culture on its way to replicative senescence, which represents the signatures of different stages of cellular senescence. These reference signatures will then be compared with gene expression signatures from human samples of different ages to estimate the proportion of cells at various senescence stages within human tissues. Since senescent cells accumulate in tissues as humans age, algorithms built on large-scale comparisons of these datasets are expected to generate a highly precise biological aging clock for humans. The Aging Clock will be very useful for our rejuvenation screening in our later phases.
We are going to apply dynamic graph modeling and structured learning on the Longitudinal Aging/Rejuvenation Atlas. Because the dataset is temporally ordered, perturbation-aware, and restricted to a single lineage, it can be naturally formulated as a time-evolving gene regulatory graph under intervention. Genes represent nodes, regulatory influences form dynamic edges, and perturbations act as structured exogenous interventions on network state transitions. This would provide a controlled setting for:
Single cell foundation model represents a convergence of big data biology and state-of-the-art AI. They have demonstrated the ability to learn from massive single-cell datasets and perform a spectrum of tasks, from labeling cells and imputing missing values to predicting experimental perturbations and uncovering gene networks. We will apply transfer learning on state-of-the-art foundation models, or Dynamic Graph ML to systematically interrogate the Longitudinal Human Aging/Rejuvenation Gene Expression Atlas.
Our strategy includes transfer learning on leading large-scale single-cell foundation models to specialize them for aging- and rejuvenation-related biological processes. Fine-tuning these models on our highly curated, longitudinally structured datasets will make them have improved signal-to-noise discrimination and reduced overfitting due to pretrained structure, and enable them to achieve superior accuracy, interpretability, and biological fidelity for downstream analyses relevant to aging biology.
These optimized models will then support in silico gene perturbation, enabling us to map causal gene–gene interactions, predict rejuvenation trajectories, and computationally screen gene combinations with unprecedented efficiency and depth.
Using the above AI-driven framework, we will achieve the following:
Upon successful preclinical validation, the optimized rejuvenation gene cocktails and their corresponding small-molecule mimetics will advance to early-stage clinical evaluation. The primary objectives will be to assess their safety and therapeutic efficacy in treating age-related diseases and, ultimately, to develop interventions capable of systemic rejuvenation and extension of human healthspan and lifespan.
Acosta-Rodríguez, V., Rijo-Ferreira, F., Izumo, M., Xu, P., Wight-Carter, M., Green, C. B., & Takahashi, J. S. (2022). Circadian alignment of early onset caloric restriction promotes longevity in male C57BL/6J mice. Science, 376(6598), 1192–1202.
Ahn, M., Kim, J., & Seo, J. H. (2025). Single-cell RNA-seq analysis reveals the multi-step process of cellular senescence. Biochemistry and Biophysics Reports, 42, 102042.
Amaral, P., Carbonell-Sala, S., De La Vega, F. M., Faial, T., Frankish, A., Gingeras, T., Guigo, R., Harrow, J. L., Hatzigeorgiou, A. G., & Johnson, R. (2023). The status of the human gene catalogue. Nature, 622(7981), 41–47.
Amorim, J. A., Coppotelli, G., Rolo, A. P., Palmeira, C. M., Ross, J. M., & Sinclair, D. A. (2022). Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nature Reviews Endocrinology, 18(4), 243–258.
Baechle, J. J., Chen, N., Makhijani, P., Winer, S., Furman, D., & Winer, D. A. (2023). Chronic inflammation and the hallmarks of aging. Molecular Metabolism, 74, 101755.
Baker, D. J., Childs, B. G., Durik, M., Wijers, M. E., Sieben, C. J., Zhong, J., A. Saltness, R., Jeganathan, K. B., Verzosa, G. C., & Pezeshki, A. (2016). Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature, 530(7589), 184–189.
Bárcena, C., Valdés-Mas, R., Mayoral, P., Garabaya, C., Durand, S., Rodríguez, F., Fernández-García, M. T., Salazar, N., Nogacka, A. M., & Garatachea, N. (2019). Healthspan and lifespan extension by fecal microbiota transplantation into progeroid mice. Nature Medicine, 25(8), 1234–1242.
Blackburn, E. H., Epel, E. S., & Lin, J. (2015). Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science, 350(6265), 1193–1198.
Bloom, S. I., Islam, M. T., Lesniewski, L. A., & Donato, A. J. (2023). Mechanisms and consequences of endothelial cell senescence. Nature Reviews Cardiology, 20(1), 38–51.
Bobkova, N. V., Evgen’ev, M., Garbuz, D. G., Kulikov, A. M., Morozov, A., Samokhin, A., Velmeshev, D., Medvinskaya, N., Nesterova, I., & Pollock, A. (2015). Exogenous Hsp70 delays senescence and improves cognitive function in aging mice. Proceedings of the National Academy of Sciences, 112(52), 16006–16011.
Boehme, M., Guzzetta, K. E., Bastiaanssen, T. F. S., van de Wouw, M., Moloney, G. M., Gual-Grau, A., Spichak, S., Olavarría-Ramírez, L., Fitzgerald, P., Morillas, E., Ritz, N. L., Jaggar, M., Cowan, C. S. M., Crispie, F., Donoso, F., Halitzki, E., Neto, M. C., Sichetti, M., Golubeva, A. V., … Cryan, J. F. (2021). Microbiota from young mice counteracts selective age-associated behavioral deficits. Nature Aging, 1(8), 666–676. https://doi.org/10.1038/s43587-021-00093-9
Browder, K. C., Reddy, P., Yamamoto, M., Haghani, A., Guillen, I. G., Sahu, S., Wang, C., Luque, Y., Prieto, J., & Shi, L. (2022). In vivo partial reprogramming alters age-associated molecular changes during physiological aging in mice. Nature Aging, 2(3), 243–253.
Campbell, K. H., McWhir, J., Ritchie, W. A., & Wilmut, I. (1996). Sheep cloned by nuclear transfer from a cultured cell line. Nature, 380(6569), 64–66.
Castoldi, F., Humeau, J., Martins, I., Lachkar, S., Loew, D., Dingli, F., Durand, S., Enot, D., Bossut, N., & Chery, A. (2020). Autophagy-mediated metabolic effects of aspirin. Cell Death Discovery, 6(1), 129.
Cohen, C., Le Goff, O., Soysouvanh, F., Vasseur, F., Tanou, M., Nguyen, C., Amrouche, L., Le Guen, J., Saltel‐Fulero, O., & Meunier, T. (2021). Glomerular endothelial cell senescence drives age‐related kidney disease through PAI‐1. EMBO Molecular Medicine, 13(11), e14146.
Cui, H., Wang, C., Maan, H., Pang, K., Luo, F., Duan, N., & Wang, B. (2024). scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nature Methods, 21(8), 1470–1480.
de Lima Camillo, L. P., Gam, R., Maskalenka, K., LeBlanc, F. J., Urrutia, G. A., Mejia, G. M., Miller, H. E., Wardlaw, C. P., Pickles, A., & Everton, L. (2025). A single factor for safer cellular rejuvenation. bioRxiv, 2025–06.
Demidenko, O., Barardo, D., Budovskii, V., Finnemore, R., Palmer III, F. R., Kennedy, B. K., & Budovskaya, Y. V. (2021). Rejuvant®, a potential life-extending compound formulation with alpha-ketoglutarate and vitamins, conferred an average 8 year reduction in biological aging, after an average of 7 months of use, in the TruAge DNA methylation test. Aging (Albany NY), 13(22), 24485.
Di Micco, R., Krizhanovsky, V., Baker, D., & d’Adda di Fagagna, F. (2021). Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nature Reviews Molecular Cell Biology, 22(2), 75–95.
Duran‐Ortiz, S., List, E. O., Ikeno, Y., Young, J., Basu, R., Bell, S., McHugh, T., Funk, K., Mathes, S., & Qian, Y. (2021). Growth hormone receptor gene disruption in mature‐adult mice improves male insulin sensitivity and extends female lifespan. Aging Cell, 20(12), e13506.
Franceschi, C., Garagnani, P., Parini, P., Giuliani, C., & Santoro, A. (2018). Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nature Reviews Endocrinology, 14(10), 576–590.
Gallardo, A., Belmonte-Reche, E., Marti-Marimon, M., Domingo-Reinés, J., Peris, G., López-Onieva, L., Fernández-Rengel, I., Xie, J., Tristán-Ramos, P., & Bellora, N. (2025). BMAL1-TRIM28 represses transposable elements independently of CLOCK in pluripotent cells. Nature Communications, 16(1), 8250.
Gilbert, S. (2000). Aging: The Biology of Senescence (6th edition.). Sunderland (MA): Sinauer Associates. https://www.ncbi.nlm.nih.gov/books/NBK10041/
Goedeke, L., Murt, K. N., Di Francesco, A., Camporez, J. P., Nasiri, A. R., Wang, Y., Zhang, X., Cline, G. W., de Cabo, R., & Shulman, G. I. (2022). Sex‐and strain‐specific effects of mitochondrial uncoupling on age‐related metabolic diseases in high‐fat diet‐fed mice. Aging Cell, 21(2), e13539.
Grosse, L., Wagner, N., Emelyanov, A., Molina, C., Lacas-Gervais, S., Wagner, K.-D., & Bulavin, D. V. (2020). Defined p16High senescent cell types are indispensable for mouse healthspan. Cell Metabolism, 32(1), 87–99.
Gurdon, J. B. (1962). Adult frogs derived from the nuclei of single somatic cells. Developmental Biology, 4(2), 256–273.
Hafycz, J. M., Strus, E., & Naidoo, N. (2022). Reducing ER stress with chaperone therapy reverses sleep fragmentation and cognitive decline in aged mice. Aging Cell, 21(6), e13598.
Hao, M., Gong, J., Zeng, X., Liu, C., Guo, Y., Cheng, X., Wang, T., Ma, J., Song, L., & Zhang, X. (2023). Large scale foundation model on single-cell transcriptomics. bioRxiv.
Hipp, M. S., Kasturi, P., & Hartl, F. U. (2019). The proteostasis network and its decline in ageing. Nature Reviews Molecular Cell Biology, 20(7), 421–435.
Jaskelioff, M., Muller, F. L., Paik, J.-H., Thomas, E., Jiang, S., Adams, A. C., Sahin, E., Kost-Alimova, M., Protopopov, A., & Cadinanos, J. (2011). Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature, 469(7328), 102–106.
Kane, A. E., & Sinclair, D. A. (2019). Epigenetic changes during aging and their reprogramming potential. Critical Reviews in Biochemistry and Molecular Biology, 54(1), 61–83.
Kiss, T., Nyúl-Tóth, Á., Balasubramanian, P., Tarantini, S., Ahire, C., DelFavero, J., Yabluchanskiy, A., Csipo, T., Farkas, E., & Wiley, G. (2020). Single-cell RNA sequencing identifies senescent cerebromicrovascular endothelial cells in the aged mouse brain. Geroscience, 42(2), 429–444.
Levine, B., & Kroemer, G. (2019). Biological functions of autophagy genes: A disease perspective. Cell, 176(1), 11–42.
Liu, S. V. (2008). iPS cells: A more critical review. Stem Cells and Development, 17(3), 391–398.
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., & Kroemer, G. (2023). Hallmarks of aging: An expanding universe. Cell, 186(2), 243–278.
Ma, S., Wang, S., Ye, Y., Ren, J., Chen, R., Li, W., Li, J., Zhao, L., Zhao, Q., & Sun, G. (2022). Heterochronic parabiosis induces stem cell revitalization and systemic rejuvenation across aged tissues. Cell Stem Cell, 29(6), 990–1005.
Maharajan, N., Vijayakumar, K., Jang, C. H., & Cho, G.-W. (2020). Caloric restriction maintains stem cells through niche and regulates stem cell aging. Journal of Molecular Medicine, 98(1), 25–37. https://doi.org/10.1007/s00109-019-01846-1
Mehdipour, M., Skinner, C., Wong, N., Lieb, M., Liu, C., Etienne, J., Kato, C., Kiprov, D., Conboy, M. J., & Conboy, I. M. (2020). Rejuvenation of three germ layers tissues by exchanging old blood plasma with saline-albumin. Aging (Albany NY), 12(10), 8790.
Miller, K. N., Victorelli, S. G., Salmonowicz, H., Dasgupta, N., Liu, T., Passos, J. F., & Adams, P. D. (2021). Cytoplasmic DNA: sources, sensing, and role in aging and disease. Cell, 184(22), 5506–5526.
Minhas, P. S., Latif-Hernandez, A., McReynolds, M. R., Durairaj, A. S., Wang, Q., Rubin, A., Joshi, A. U., He, J. Q., Gauba, E., & Liu, L. (2021). Restoring metabolism of myeloid cells reverses cognitive decline in ageing. Nature, 590(7844), 122–128.
Nieto-Torres, J. L., & Hansen, M. (2021). Macroautophagy and aging: The impact of cellular recycling on health and longevity. Molecular Aspects of Medicine, 82, 101020.
North, B. J., Rosenberg, M. A., Jeganathan, K. B., Hafner, A. V., Michan, S., Dai, J., Baker, D. J., Cen, Y., Wu, L. E., & Sauve, A. A. (2014). SIRT 2 induces the checkpoint kinase BubR1 to increase lifespan. The EMBO Journal, 33(13), 1438–1453.
Ocampo, A., Reddy, P., Martinez-Redondo, P., Platero-Luengo, A., Hatanaka, F., Hishida, T., Li, M., Lam, D., Kurita, M., & Beyret, E. (2016). In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell, 167(7), 1719–1733.
Olova, N., Simpson, D. J., Marioni, R. E., & Chandra, T. (2019). Partial reprogramming induces a steady decline in epigenetic age before loss of somatic identity. Aging Cell, 18(1), e12877.
Pálovics, R., Keller, A., Schaum, N., Tan, W., Fehlmann, T., Borja, M., Kern, F., Bonanno, L., Calcuttawala, K., & Webber, J. (2022). Molecular hallmarks of heterochronic parabiosis at single-cell resolution. Nature, 603(7900), 309–314.
Pyo, J.-O., Yoo, S.-M., Ahn, H.-H., Nah, J., Hong, S.-H., Kam, T.-I., Jung, S., & Jung, Y.-K. (2013). Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nature Communications, 4(1), 2300.
Ragonnaud, E., & Biragyn, A. (2021). Gut microbiota as the key controllers of “healthy” aging of elderly people. Immunity & Ageing, 18(1), 2. https://doi.org/10.1186/s12979-020-00213-w
Ruzankina, Y., & Brown, E. (2007). Relationships between stem cell exhaustion, tumour suppression and ageing. British Journal of Cancer, 97(9), 1189–1193.
Sciorati, C., Gamberale, R., Monno, A., Citterio, L., Lanzani, C., De Lorenzo, R., Ramirez, G. A., Esposito, A., Manunta, P., & Manfredi, A. A. (2020). Pharmacological blockade of TNFα prevents sarcopenia and prolongs survival in aging mice. Aging (Albany NY), 12(23), 23497.
Shosha, E., Xu, Z., Narayanan, S. P., Lemtalsi, T., Fouda, A. Y., Rojas, M., Xing, J., Fulton, D., Caldwell, R. W., & Caldwell, R. B. (2018). Mechanisms of diabetes-induced endothelial cell senescence: Role of arginase 1. International Journal of Molecular Sciences, 19(4), 1215.
Takahashi, K., & Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell, 126(4), 663–676.
Tavallaie, M., Voshtani, R., Deng, X., Qiao, Y., Jiang, F., Collman, J. P., & Fu, L. (2020). Moderation of mitochondrial respiration mitigates metabolic syndrome of aging. Proceedings of the National Academy of Sciences, 117(18), 9840–9850.
Theodoris, C. V., Xiao, L., Chopra, A., Chaffin, M. D., Al Sayed, Z. R., Hill, M. C., Mantineo, H., Brydon, E. M., Zeng, Z., & Liu, X. S. (2023). Transfer learning enables predictions in network biology. Nature, 618(7965), 616–624.
Tian, X., Firsanov, D., Zhang, Z., Cheng, Y., Luo, L., Tombline, G., Tan, R., Simon, M., Henderson, S., & Steffan, J. (2019). SIRT6 is responsible for more efficient DNA double-strand break repair in long-lived species. Cell, 177(3), 622–638.
Tomas-Loba, A., Flores, I., Fernandez-Marcos, P. J., Cayuela, M. L., Maraver, A., Tejera, A., Borras, C., Matheu, A., Klatt, P., & Flores, J. M. (2008). Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell, 135(4), 609–622.
Vijg, J., & Suh, Y. (2013). Genome instability and aging. Annual Review of Physiology, 75(1), 645–668.
Wang, T., Li, Y., Zhu, Y., Liu, Z., Huang, L., Zhao, H., Zhou, Z., & Wu, Q. (2023). Anti-aging mechanism of different age donor-matched adipose-derived stem cells. Stem Cell Research & Therapy, 14(1), 192. https://doi.org/10.1186/s13287-023-03415-3
Wang, W., Zheng, Y., Sun, S., Li, W., Song, M., Ji, Q., Wu, Z., Liu, Z., Fan, Y., & Liu, F. (2021). A genome-wide CRISPR-based screen identifies KAT7 as a driver of cellular senescence. Science Translational Medicine, 13(575), eabd2655.
Xu, M., Pirtskhalava, T., Farr, J. N., Weigand, B. M., Palmer, A. K., Weivoda, M. M., Inman, C. L., Ogrodnik, M. B., Hachfeld, C. M., & Fraser, D. G. (2018). Senolytics improve physical function and increase lifespan in old age. Nature Medicine, 24(8), 1246–1256.
Yang, F., Wang, W., Wang, F., Fang, Y., Tang, D., Huang, J., Lu, H., & Yao, J. (2022). scBERT as a large-scale pretrained deep language model for cell type annotation of single-cell RNA-seq data. Nature Machine Intelligence, 4(10), 852–866.
Yang, J.-H., Hayano, M., Griffin, P. T., Amorim, J. A., Bonkowski, M. S., Apostolides, J. K., Salfati, E. L., Blanchette, M., Munding, E. M., & Bhakta, M. (2023). Loss of epigenetic information as a cause of mammalian aging. Cell, 186(2), 305–326.
Yokoi, T., Fukuo, K., Yasuda, O., Hotta, M., Miyazaki, J., Takemura, Y., Kawamoto, H., Ichijo, H., & Ogihara, T. (2006). Apoptosis signal-regulating kinase 1 mediates cellular senescence induced by high glucose in endothelial cells. Diabetes, 55(6), 1660–1665.
Yu, J., Vodyanik, M. A., Smuga-Otto, K., Antosiewicz-Bourget, J., Frane, J. L., Tian, S., Nie, J., Jonsdottir, G. A., Ruotti, V., & Stewart, R. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science, 318(5858), 1917–1920.
Zeng, Y., Xie, J., Shangguan, N., Wei, Z., Li, W., Su, Y., Yang, S., Zhang, C., Zhang, J., & Fang, N. (2025). CellFM: a large-scale foundation model pre-trained on transcriptomics of 100 million human cells. Nature Communications, 16(1), 4679.
Dr. Gordon Ma is a geneticist, stem cell biologist, and biotech founder with over 20 years of experience in epigenetics, gene regulation, and rejuvenation biology. His PhD work produced the first evidence of CDH1 promoter methylation in precancerous tissue—a foundational contribution to modern epigenetics. He completed postdoctoral training at Harvard Medical School and the University of Colorado, where he studied XIST-mediated epigenetic regulation.
At the NIH (NHLBI), Dr. Ma conducted research in embryonic stem cell differentiation and cardiovascular regenerative biology and made key findings on mitochondrial regulation of pluripotency. He later founded several life science companies while continuing NIH research as a special volunteer for nearly a decade.
As Founder and CEO of Rejuve Therapeutics, Dr. Ma is the principal inventor of a patented method demonstrating gene-driven rejuvenation without dedifferentiation. His ongoing work includes large-scale single-cell sequencing studies and the development of the first Longitudinal Human Aging and Rejuvenation Gene Expression Atlas. He is collaborating closely with AI scientists on transfer learning and biological foundation models to identify key regulatory genes driving aging and rejuvenation and to develop clinically viable rejuvenation gene cocktails